
歐盟對第12條規定的醫療器械系統和程序包的規定
第12條是什么?
1.當公司將各自帶有CE標志的醫療設備組合在一起并且根據CE認證標志打算使用產品時,該組件被稱為“系統”或“程序包”。出于本文的目的,我們將組件稱為“系統”。
2.醫療器械指令(MDD)93/42 / EEC第12條定義了將各個醫療器械配置到系統中所需的合格評定,并確保在將這些系統投入使用和使用時的安全性。
3.對于第12條的適用,構成系統的每個醫療器械必須具有自己的CE標志。
4.第12條通常適用于將來自多個制造商和不同的認證機構(如果適用)的設備組裝在一起形成一個單元的情況。當制造商僅將自己的設備組合到一個系統中時,這些設備屬于第11條要求,而第12條很可能不需要考慮。但是,匯編程序最終有責任確定第12條對設備的適用性。
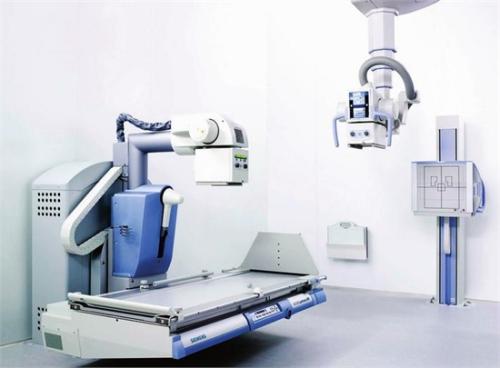
系統匯編程序的職責
1.系統組裝者負責驗證設備的兼容性,包裝系統,向用戶提供相關信息以及在適當的內部控制和檢查方法下執行組裝。匯編程序必須能夠提供已簽署的聲明,表明這些責任已得到滿足。
2.作為合法制造商,匯編人員向歐盟主管部門負責報告。這包括系統在歐盟成員國的主管當局注冊,其中匯編人或其授權代表成立。
驗證相互兼容性
1.根據第12條的規定,系統組裝者必須驗證單獨CE標記設備的“相互兼容性”。設備的相互兼容性必須基于原始設備制造商規定的每個設備的預期用途和使用限制。在組裝系統時,各個設備的預期用途不能改變原始制造商指定的范圍。
2.系統組裝者聲明,基于系統的每個組件(醫療設備)可用的使用說明,這些設備是兼容的并且能夠在組裝時一起工作,以確保系統的安全和正確使用。該聲明還暗示各個設備的指定性能未受損。
非醫療器械
如果系統中的產品不是所有醫療設備怎么辦?應評估每個組件,以了解每個組件的法規要求。并且,必須為產品獲得適當的法規遵從性。例如,如果系統包含藥品,那些產品必須符合藥品指令2001/83 / EC,并且必須相應標記產品。風險評估應記錄這些藥品應按醫藥產品制造商的意圖使用,并提供相關的使用信息。
打包您的系統并提供相關信息
1.裝配商必須根據各個設備的原始制造商規定的規格對其系統進行打包。這些規范包括為各個設備建立的預期用途和使用限制。此外,規范定義了CE標記設備可以組裝并作為系統投放市場的條件。
2.系統的組裝者應始終確保每個設備都帶有CE標志,并且原始制造商已獲得適當的批準。要求每個原始設備制造商提供MDD符合性聲明以及(如果適用)提供根據MDD第11條規定的適當程序進行評估的客觀證據的公告機構證書符合匯編程序的最佳利益。
3.重要的是要注意,在最終組裝的系統中,每個單獨的醫療設備保持并具有其自己的CE標記; 但是,組裝好的系統本身不帶CE標志。
4.如果裝配商在整個系統中貼上CE標志,這意味著符合第11條和公告機構的審查,假設設備的風險高于I級自我認證。
5.系統也必須附帶有關系統安全和正確使用的相關信息。此信息必須符合MDD附件I(13)中規定的要求,并在適當情況下包括原始設備制造商提供的信息。
在內部控制和檢查下組裝
匯編人必須聲明第12條程序的“整體活動”已經過適當的內部控制和檢查方法。定義控制和檢查的哪種方法適用于第12條系統以及這些方法如何被認為對系統“適當”符合組裝者的最佳利益。
符合性聲明
1.必須生成并簽署符合第12條的聲明,說明匯編人員具有:
2.根據制造商的說明驗證設備的相互兼容性,并按照說明進行操作; 和
3.包裝系統或程序包,并向包含制造商相關說明的用戶提供相關信息; 和
4.整個活動采用適當的內部控制和檢查方法。
5.所有第12條聲明必須由主管當局保管五年。
總結:考慮點
1.當系統包含不帶有自己的CE標志的設備時,該系統不屬于第12條,必須遵守MDD第11條的相關程序。
2.如果設備組合與其原始用途不相符,則該系統不屬于第12條,必須遵守第11條規定的相關程序。
3.即使第12條可以應用于特定系統,裝配工也可以選擇將系統本身視為一種裝置,并根據MDD第11條采用相關程序。如果該系統將受第11條的保護,則該系統必須擁有公告機構頒發的CE標志證書。
4.第12條的目的是提供簡化的監管機制,以便可以組裝受到適當監管合規的醫療器械組件,主要是為了方便(向FDA借用一個術語)給用戶。